大环类的天然产物现已成为药物先导化合物发现的重要来源之一。由于其具有独特的环状骨架,3D构象受限,适当的刚性和柔性等特点,大环化策略在药物设计中备受青睐。然而,受自然界生物合成途径的制约,天然的大环化合物仍然存在着结构多样性不足等问题,从而不可避免地限制了大环类药物的研发。因此,我们急需发展新的方法来设计合成结构丰富和生物活性多样的类天然大环化合物,从而加速大环先导化合物的发现。
此前,上海药物研究所的杨伟波课题组通过仿生模块化的设计策略成功构建了一系列具有生物学活性的大环化合物库。在这里,他们以天然产物中广泛存在的苯基吡啶结构和含有α-芳基苯乙酮的环状结构为基础,设计了如下图所示的新型的类天然大环化合物,并通过发展一种新的C-H/O2双活化的反应,将原本所需的三步的传统反应缩短为一步,从而极大地加速此类大环化合物的合成。这一反应突破了原本C-H/O2双活化反应只限于两组分的局限性,为后阶段闭环反应提供了新的反应类型。表型筛选发现了部分大环化合物具有较好的抗病毒H1N1活性。
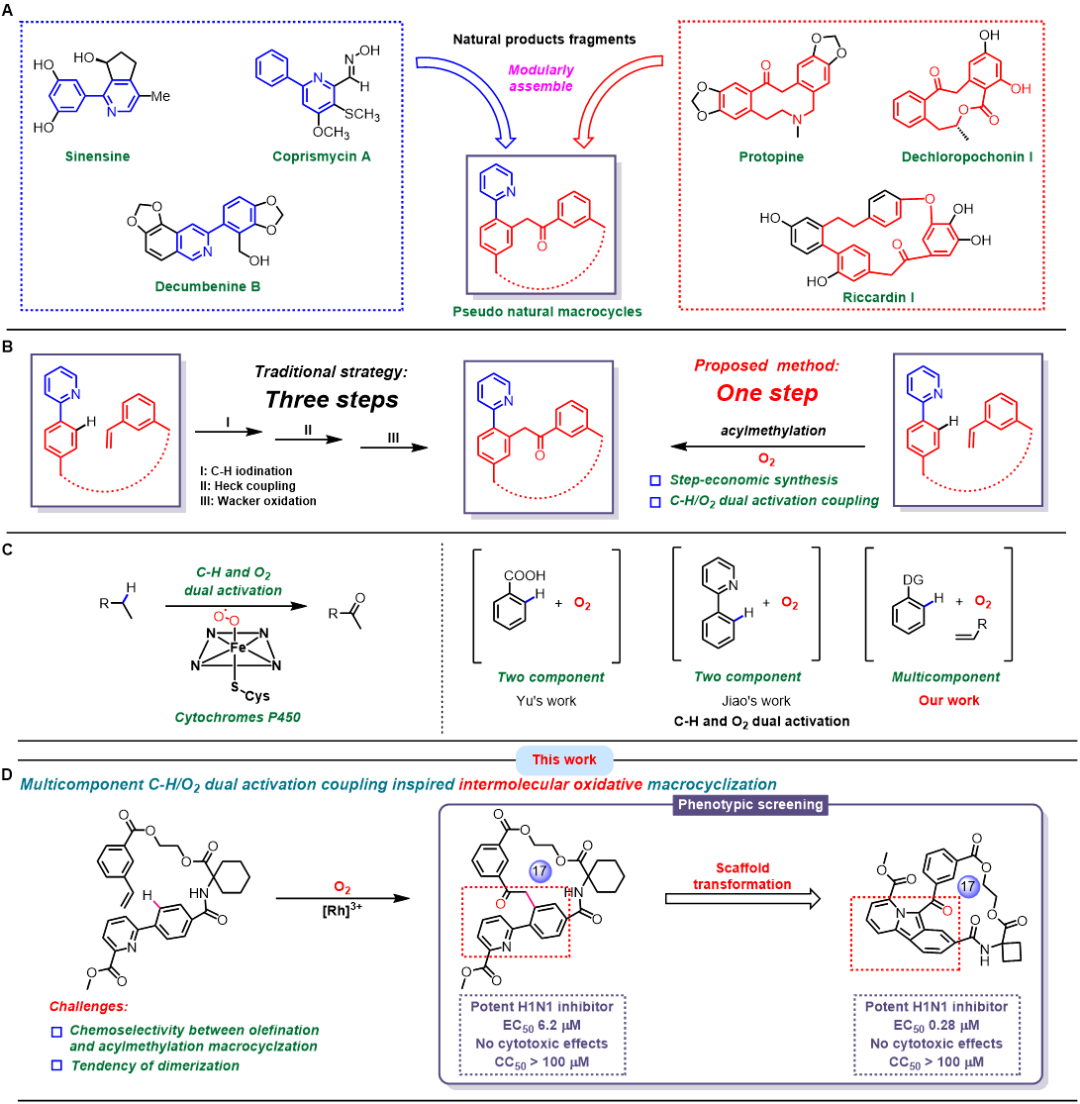
图1:仿生模块设计大环与铑催化C-H/O2双活化大环化方法的发现
首先,他们通过以分子间反应为模板反应,成功筛选得到了最佳的反应条件,并且这一反应条件对于复杂药物分子的后阶段修饰同样可以兼容。此外,这一反应条件可以直接应用于后阶段的大环化合物的合成,对于不同的天然和非天然的氨基酸(包括螺环、桥环等片段)均可以得到中等至良好的产率。值得注意的是,反应生成的大环化合物可进一步在铜催化下转化为结构新颖的氮杂稠环化合物。
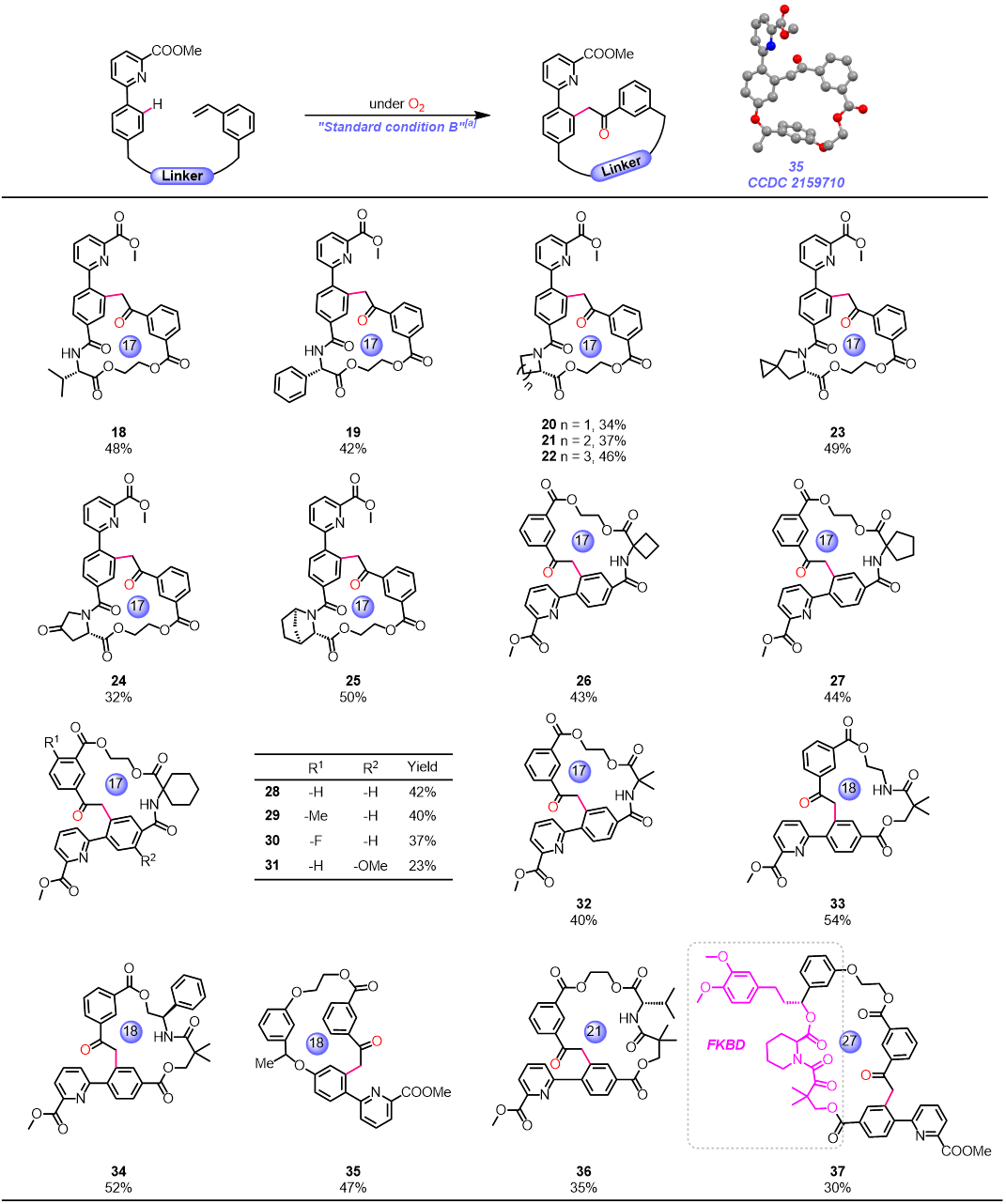
图2:铑催化C-H/O2双活化大环化构建结构多样的大环库
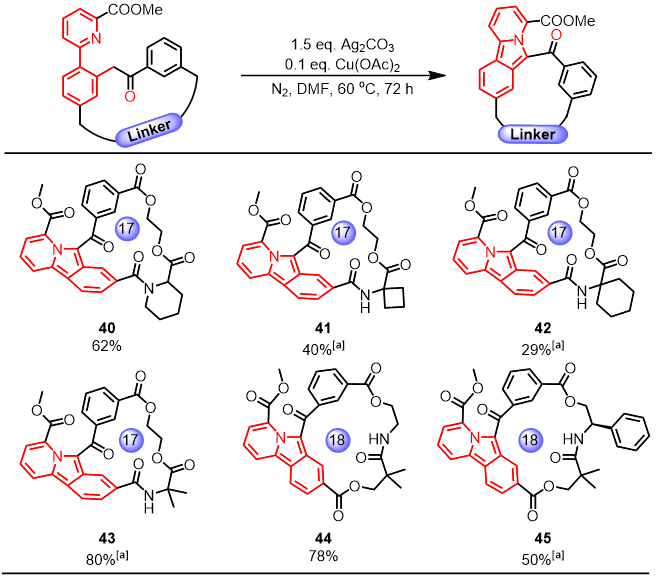
图3:铑催化C-H/O2双活化大环化构建结构多样的大环分子库
通过活性筛选,作者发现合成得到的含有α-芳基苯乙酮结构和氮杂稠环结构的大环化合物具有较好的抗病毒H1N1活性,进一步构效关系研究表明氮杂稠环结构的大环活性最佳,其EC50值为0.28 μM,从而为抗病毒H1N1的大环类药物的开发提供了重要的参考。最后,他们还通过同位素标记实验、对照试验、竞争性实验、DFT计算等方式,对反应的机理进行了深入的研究,揭示了反应的决速步为O-O键断裂的过程,为以后的反应设计新骨架提供了新的指导。
总之,该研究工作把合成方法学与药物化学进行了有效交叉融合,从源头上构建全新骨架的大环化合物,不仅为方法学提供了新的应用方向,而且为药物化学的发展带来了重要的技术支撑和科学指导。
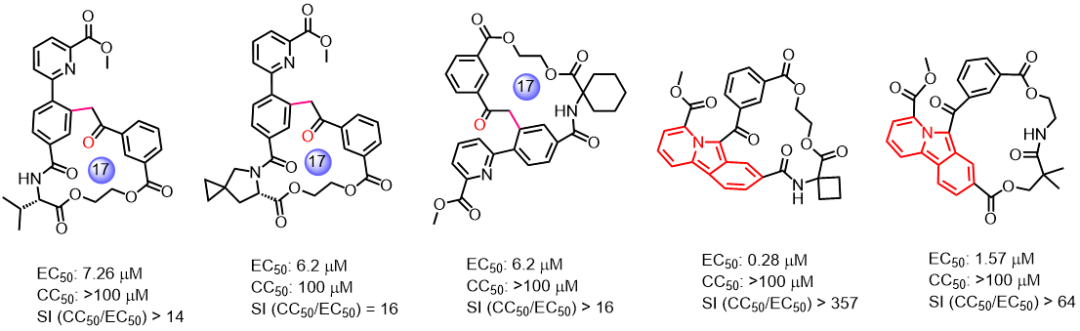
图4:抗病毒H1N1的大环先导化合物发现
以上文章转载于微信公众号 WileyChem ,如有侵权,请及时联系我们修改或进行删除。
